Williams Endocrinology 15e Ch33|第 2 型糖尿病的病理生理
KEY TAKEAWAYS
- T2D 是全球最常見的糖尿病型態,佔 90–95%。IDF 估 2021 年全球 5.37 億成人罹病,2045 年將升至 7.38 億;美國 CDC 估 11.3% 美國成人為糖尿病、38% 為糖尿病前期。
- T2D 的病理生理具四大核心異常:周邊組織(肌肉、脂肪、肝臟)的胰島素阻抗、葡萄糖刺激下的胰島素分泌缺陷、肌肉葡萄糖攝取下降、肝臟葡萄糖製造增加。額外特徵包含高升糖素血症、incretin 分泌或作用異常、脂解加速、腎小管再吸收增加、CNS 代謝調控異常。
- 在預先傾向 T2D 的個體中,胰島素阻抗早於高血糖出現可達 20 年以上;β 細胞功能缺陷亦於糖尿病前期即可偵測,故二者並存於臨床發病時。
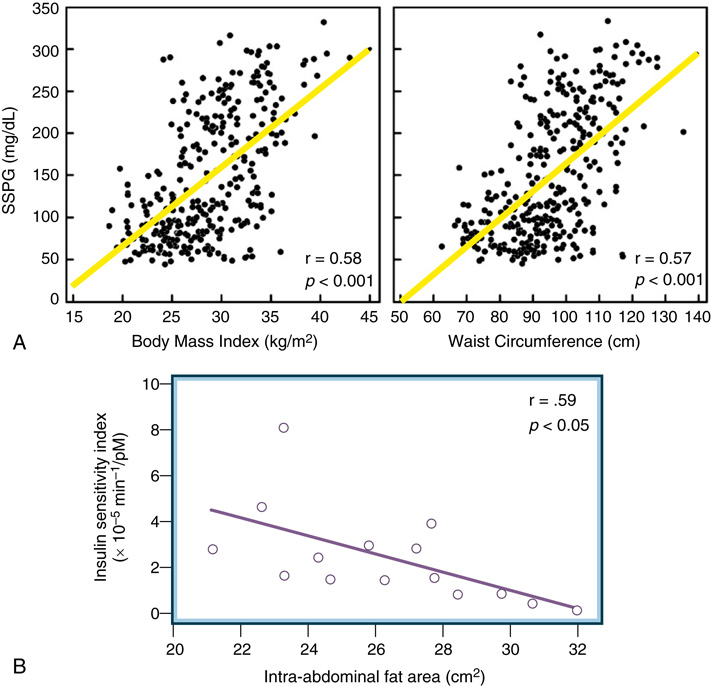
- 中央型(內臟)肥胖比皮下脂肪更強烈關聯胰島素阻抗。亞裔(含東南亞)於 BMI ≥ 23 kg/m² 即建議篩檢,因內臟脂肪比例較高。
- 兒童/青少年 T2D 比例正快速攀升(美國 10–19 歲 youths 由 2001 年 3.4/萬升至 2017 年 6.7/萬,相對增加 95%),西班牙裔與非西班牙裔黑人增幅尤大。
- 單基因型糖尿病(insulin receptor 突變、Donohue / Rabson-Mendenhall syndrome、generalized 與 partial lipodystrophy)罕見但需早期辨識;可決定治療策略。Insulin receptor 突變者可能需 > 10,000 U/day insulin。
- 多基因型 T2D 已找到 700+ 個關聯基因,總體解釋遺傳風險 ≤ 10%;TCF7L2 是常見變異中 odds ratio 最大者;KCNJ11/ABCC8 突變所致的新生兒糖尿病可用 sulfonylurea 取代 insulin。
- 表觀遺傳:母體子宮內過度或不足營養(如荷蘭飢餓冬天 1944–45)的後代肥胖風險升高,效應可跨代延續。
- 胰島素訊息透過 IRS-PI3K-AKT 三節點主導代謝;組織特異性表現使同一受體在肌肉(GLUT4 移位、蛋白合成)、脂肪(脂肪生成、抑脂解)、肝(脂肪生成、抑糖質新生)展現迥異的代謝結果。
- “Non-classical” insulin-sensitive tissues(β cell、α cell、內皮、心肌、巨噬細胞、CNS)的胰島素訊息缺失與 atherosclerosis、metabolic syndrome、Alzheimer disease 相關。
- 胰島素阻抗的細胞機轉:serine/threonine 磷酸化抑制 IR 與 IRS、PTP1B 與 LAR 增加去磷酸化、營養過載活化 mTOR/S6K、JNK、IKK 形成 feedback inhibition、ER stress / UPR、innate immunity 活化、粒線體功能下降、脂肪組織衍生 sEV 透過 miRNA 干擾標的組織。
- Ectopic lipid(肌內、肝內 triglyceride)伴隨 DAG 與 ceramide 累積,活化 novel PKC isoforms (PKCθ/δ),干擾 IRS1 訊息;運動訓練即使讓肌內 triglyceride 增加亦不致胰島素阻抗,因 perilipin (PLIN2/3/5) 包覆隔離脂滴。
- Hepatic insulin resistance 對空腹高血糖貢獻量級與周邊葡萄糖處置缺損相當或更大;metformin 透過抑制肝臟糖製造改善 glucose tolerance。
- 晝夜節律破壞(睡眠不足、輪班、阻塞型睡眠呼吸中止)為獨立的 T2D 風險;time-restricted eating(6–10 小時進食窗)可改善胰島素敏感性與血壓,部分益處需要伴隨體重下降。
- 腸道菌群透過短鏈脂肪酸、膽酸代謝、屏障功能、內毒素血症介導胰島素阻抗;prebiotics、probiotics、糞菌移植是潛在治療途徑但仍需驗證。
- 特殊狀況導致胰島素阻抗:妊娠(胎盤生長素、胎盤泌乳素)、glucocorticoids(IRS1 下調+肝糖製造增加)、calcineurin inhibitors(主要降低 insulin 分泌)、mTOR inhibitors(直接抑制下游訊息)、HIV protease inhibitors、glucotoxicity(hexosamine pathway 過載)、術後高血糖(epinephrine 介導)、statins、antipsychotics(olanzapine、clozapine 增重最甚)。
流行病學|Epidemiology
- T2D 是全球糖尿病的主要型態,佔病例 90–95%。
- 開發中與已開發國家同步盛行。
- IDF(International Diabetes Federation)估 2021 年全球 20–79 歲糖尿病人 5.37 億 → 2045 年將達 7.38 億。
- 目前以高所得國家為多,但新增病例最大成長將出現於中低所得國家(人口成長較大)。
- 美國 CDC 2022 年估:3,730 萬人(人口 11.3%)罹病,其中 850 萬人(23.0%)未確診;以 fasting glucose 或 HbA₁c 標準另有 9,600 萬人(成年 38%)為糖尿病前期。
- 經濟負擔
- 2021 年 IDF 估全球糖尿病相關健康支出 9,660 億美元。
- ADA 估美國 2017 年直接醫療支出 2,370 億美元、生產力損失 900 億美元;按年齡性別校正,糖尿病人均支出為非糖尿病者的 2.3 倍。
- 美國高昂藥價(含 insulin)使許多病人無法依處方取得藥物,導致併發症與經濟負擔加劇。
- T2D 風險因子總結於 Box 33.1。
- T2D 發生於遺傳易感者暴露於誘發臨床發病的環境因素。
- 雖與高 BMI 強烈關聯,但脂肪分布與所需過量脂肪在不同族群差異大。
- 內臟脂肪(中央型肥胖)多者比皮下脂肪多者更具胰島素阻抗、T2D 風險更高。
- 中央肥胖伴隨肝、肌肉的 ectopic fat 累積,加劇胰島素阻抗。
- 東南亞族群於較低 BMI 即可發 T2D(內臟脂肪比例較高)→ 建議亞裔 BMI ≥ 23 kg/m² 即啟動 T2D 篩檢。
- 移民效應:易感族群移居美國採西式飲食 → 體重上升、T2D 速增;亞洲西化飲食擴散後,本地 T2D 增加,移民效應消失。
- 美國族群差異:原住民 14.5% > 非西班牙裔白人 7.4%(圖 33.1)。
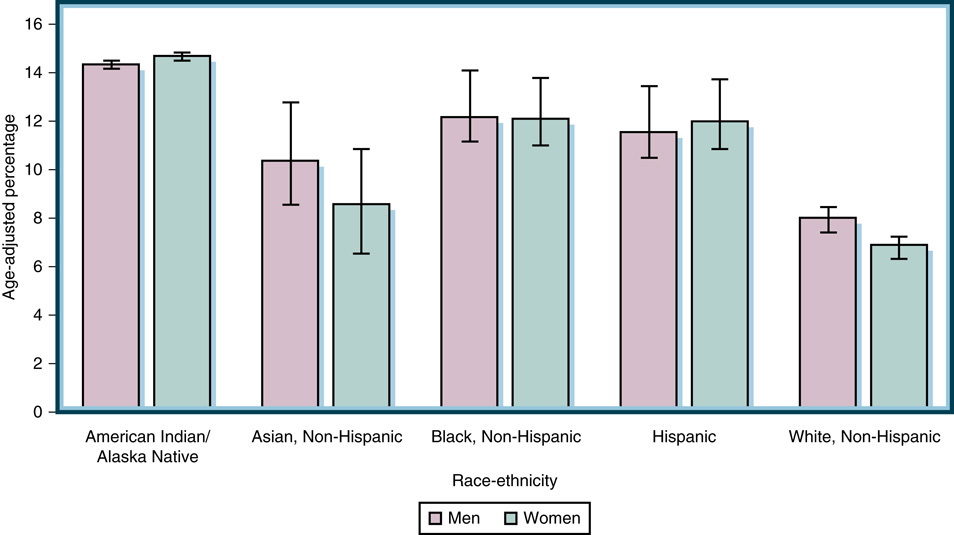
Box 33.1 T2D 流行病學決定因子與風險因子
- 遺傳因素:基因標記、家族病史。
- 人口學:年齡、族群。
- 行為與生活型態:肥胖(含分布、持續時間)、缺乏運動、飲食、壓力、西化/都市化/現代化、藥物、輪班工作。
- 代謝決定因子與中間風險:葡萄糖耐受性異常、胰島素阻抗、妊娠糖尿病、母親妊娠糖尿病的後代、子宮內營養不足或過剩、菌群組成。
- 兒童/青少年 T2D 上升
- 過去兒童糖尿病以 T1D 為主(T2D 僅 1–2%),現美國新診斷兒童糖尿病近 30% 為 T2D。
- 10–19 歲 youths T2D 盛行率:2001 年 3.4/萬 → 2017 年 6.7/萬,16 年相對增加 95%。
- 西班牙裔與非西班牙裔黑人增幅尤大(5.7 與 8.5/萬)。
- 罕見的單基因糖尿病也愈來愈被辨識。
致病機轉|Pathogenesis
- T2D 致病涉及遺傳、表觀遺傳、環境因子的交互作用。
- 主要環境因子:熱量過量導致肥胖、久坐生活型態(圖 33.2)。
- 其他環境因子:藥物、發炎、晝夜節律破壞、菌群。
- 臨床表現異質性大:發病年齡、高血糖嚴重度、肥胖程度均有寬廣分布。
- 病理生理上的四大核心異常:
- 周邊組織(肌肉、脂肪、肝臟,胰島素作用之經典組織)的胰島素阻抗。
- 對葡萄糖刺激的胰島素分泌缺陷(雖絕對值可高、可低、可正常)。
- 肌肉葡萄糖攝取下降 → 餐後高血糖。
- 肝臟葡萄糖製造增加 → 空腹高血糖。
- 額外特徵:高升糖素血症、incretin 分泌或作用改變、脂肪細胞脂解加速、腎小管再吸收增加、CNS 代謝調控異常。
- 胰島素阻抗 vs β 細胞缺陷的相對重要性仍存爭議
- 已知最早可偵測異常為胰島素阻抗,可早於 T2D ≥ 20 年。
- 諸多狀況進一步加劇胰島素阻抗、加重 β 細胞分泌負擔:青春期、妊娠、久坐生活、體重增加。
- GWAS 鑑出多型性提示 β 細胞失能的遺傳易感,但無單一基因或單一檢驗能預測結局。
第 2 型糖尿病發展中的遺傳因子|Genetic Factors in the Development of Type 2 Diabetes Mellitus
- 遺傳、環境、病理生理因子互動方式複雜,因人而異。
- 常見 T2D 為多基因型,由多基因 × 多重環境因子(已知與未知)× 表觀遺傳因子互動所致。
- 因此個別基因難辨識、貢獻度小。
- 單基因型雖罕見但重要:揭示正常生理機制,早期辨識可決定適切治療(見 Ch37)。
與胰島素阻抗相關的單基因型糖尿病|Monogenic Forms of Diabetes Associated With Insulin Resistance
- 單基因型中該基因為 necessary and sufficient;環境因子幾乎無關,但同一突變的個體外顯率變異大。
- 通常於前 2–3 個十年診斷;若僅輕度無症狀高血糖,可能延後診斷。
- 一項研究:自詡 T1D ≥ 50 年者,基因型化發現近 8% 帶單基因型糖尿病突變。
- 機轉分為 insulin secretion 缺陷型 vs insulin response/resistance 缺陷型;本章只談後者。
Insulin Receptor 突變|Mutations in the Insulin Receptor
- Insulin receptor 基因已鑑定多種突變,至少三個臨床症候群:
- Type A insulin resistance:胰島素阻抗、acanthosis nigricans、hyperandrogenism;多於青春期或年輕成人診斷,常因 acanthosis nigricans 或男性化徵兆而非血糖異常被發現;可能伴隨嚴重高胰島素血症。
- Donohue syndrome(舊稱 leprechaunism):嚴重子宮內生長遲滯、特殊面容(leprechaunism)、出生 1–2 年內死亡;雖嚴重胰島素阻抗,但因極高 insulin 可有空腹低血糖。
- Rabson-Mendenhall syndrome:身材矮小、腹部膨出、牙齒/指甲異常,部分患者有 pineal hyperplasia。
- 突變部位影響表型:
- Type A:多在 intracellular tyrosine kinase domain。
- Donohue 與 Rabson-Mendenhall:多在 extracellular ligand-binding domain 或 FnIII(影響受體摺疊)。
- 臨床特徵:嚴重高胰島素血症 + acanthosis nigricans(即使無肥胖)。
- 部分患者因內生 insulin 大量代償可維持正常血糖;部分需 > 10,000 U/day insulin 仍無反應。
脂質代謝障礙性糖尿病|Lipodystrophic Diabetes
- 可為遺傳或獲得性;特徵為嚴重胰島素阻抗 + 脂肪缺失(lipoatrophy)/ 脂肪錯誤分布(lipodystrophy)。
- 臨床:皮下脂肪減少、胰島素阻抗、高三酸甘油酯血症。
- 遺傳型分為 generalized 與 partial 兩類;多種致病基因參與不同細胞功能:
- Insulin signaling: AKT2, PIK3R1
- Caveolins: CAV1, PTRF
- Phospholipid biosynthesis: AGPAT2, PCYT1A
- Lipid droplet morphology: LMNA, BSCL2, ZMPSTE24
- Adipogenesis: CIDEC, PPARG
- Lipolysis: LIPE, PLIN1
- LMNA 同基因可因不同突變致 face-sparing familial partial lipodystrophy(Dunningan syndrome),或 mandibuloacral dysplasia syndrome(伴生長遲滯與顱面骨骼畸形)。
- Generalized lipodystrophy 罕見但臨床易辨:全身皮下脂肪明顯缺失。
- 嚴重代謝異常(嚴重脂肪肝、ascites、esophageal varices)。
- leptin 與 adiponectin 極低。
- Leptin replacement 改善血糖、脂肪肝、三酸甘油酯。
- Partial lipodystrophy 常被忽略,真正發生率不明。
- 表型隨突變與性別差異大。
- leptin 多正常或略低,多數 leptin replacement 無效。
- Acquired generalized lipodystrophy(Seip-Lawrence syndrome):兒童/青少年/年輕成人發病,由臉、手臂、腿開始;推測為自體免疫破壞脂肪組織,但決定性證據缺乏。
多基因型 T2D 的遺傳學|Genetics of the Polygenic Forms of Type 2 Diabetes Mellitus
- 病理複雜:肌肉、脂肪、肝臟胰島素阻抗 + β 細胞分泌缺陷 → 葡萄糖攝取下降 + 肝糖製造增加。
- 雖家族病史強,但決定病程的主要缺陷仍未明確;> 700 個基因關聯 T2D,個別基因貢獻與基因–環境互動仍未明。
- 胰島素阻抗在易感者於高血糖前即存在,提示為主要異常之一。
- 部分嚴重胰島素阻抗者可極端高胰島素分泌作補償;提示輕度胰島素阻抗者亦應能補償。
- 因此,胰島素阻抗 + 部分 β 細胞缺陷可能皆於糖尿病前期就已存在。
- 一級親屬中,葡萄糖正常者已可見胰島素分泌異常,支持 β 細胞缺陷與胰島素阻抗並存。
- 結論:何者為「主要」缺陷仍爭論,但臨床發病時通常二者並存。
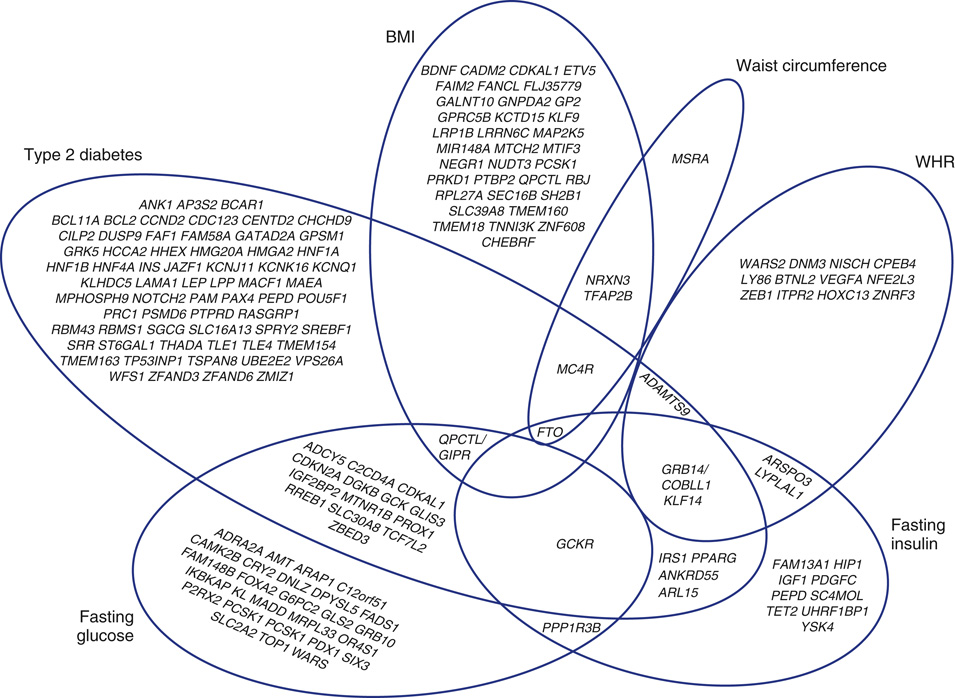
- GWAS 進展:先前候選基因法或 linkage 法 → 近年 GWAS 不偏式比較 cases vs controls 找出 SNP 關聯,但 SNP 多位於非編碼基因內區,致病基因不易確認(圖 33.3)。
Insulin Receptor Substrate 1 基因|Insulin Receptor Substrate 1 Gene
- T2D 第一個鑑出的多型性:IRS-1 的 Gly972Arg。
- 透過直接定序 86 名 T2D + 76 對照,cases 出現率為 controls 的 3 倍。
- 機轉:減弱 insulin-stimulated PI3K signaling。
- 隨後資料顯示其在 T2D 整體風險中相對小,但為 T2D 與遺傳關聯的首例。
Transcription Factor 7-Like 2 基因|Transcription Factor 7–Like 2 Gene
- Grant 等於冰島 T2D 與 controls 中以 228 個 microsatellite 標記基因型化,發現 TCF7L2(舊稱 TCF4)內隆 3 的 microsatellite DG10S478 與 T2D 關聯;於丹麥與美國 cohort 重現。
- 攜帶 at-risk allele 的雜合與同合子(38% 與 7%)相對風險為 1.45 與 2.41。
- 進一步研究:特定 TCF7L2 多型性增加 IGT → T2D 進展風險,並降低 glucose-stimulated insulin secretion。
- TCF7L2 為 high-mobility group box 轉錄因子,原本與大腸癌關聯。
- 提議機轉:透過 Wnt signaling 調控 adipogenesis 與腸道內分泌細胞 proglucagon 表現。
- 在常見變異中,TCF7L2 對 T2D 影響最大;但多型性多為 intronic,是否影響蛋白功能仍未明。
KATP 通道基因 KCNJ11 與 ABCC8|KATP Channel Genes: KCNJ11 and ABCC8
- β 細胞 KATP 通道由 Kir6.2(KCNJ11)+ SUR1(ABCC8)構成。
- Kir6.2 為孔道;SUR1 為 sulfonylurea 受體,控制通道開啟。
- 通道開 → 鉀外流 → insulin 分泌。
- 突變為新生兒糖尿病最常見原因;辨識重要,因可用 sulfonylurea 治療而非 insulin。
- 兩基因位於 11 號染色體相鄰位置,皆有 SNP 關聯成人 T2D。
- KCNJ11 missense Glu23Lys(E23K)增加成人 T2D 風險平均 13%,KK 同合子相對風險 1.28。
- 一研究提示這類多型性導致的成人 T2D,亦可能對 sulfonylurea 反應較佳。
PPARγ 基因|Peroxisome Proliferator–Activated Receptor γ Gene
- PPARγ 為核受體,調控脂質與葡萄糖恆定、細胞分化。
- 主要表達於脂肪,亦在肌肉與 β 細胞。
- 肌肉 PPARγ 剔除 → 脂肪酸氧化異常;β 細胞剔除 → 高脂飲食下 β 細胞質量增加變鈍。
- 常見 missense Pro12Ala(P12A)於 PPARG(編碼 PPARγ2):alanine allele 風險比 0.79。
- Pro12 同合子比攜帶 Ala12 者多 1.25 倍 T2D 風險。
- 第二個多型性 C161 → T 在西班牙裔與非西班牙裔白人女性與胰島素阻抗關聯。
- PPARγ loss-of-function 突變致 familial partial lipodystrophy type 3(FPLD3),TZD 治療反應顯著。
HNF4α 基因|Hepatocyte Nuclear Factor 4α Gene
- HNF4A 是首發現的 MODY 基因,造成胰島素分泌異常。
- HNF4α 亦在肝臟透過替代啟動子表達,調控糖質新生。
- 帶風險 allele 者:肝糖製造增加,而非 insulin 分泌缺陷,符合替代肝臟啟動子的多型性。
- 多項臨床研究確認該肝臟特異性啟動子 HNF4A 的遺傳變異增加 T2D 風險。
Kruppel-Like Factor 14(KLF14)
- KLF14 為母系印記轉錄因子。
- 非編碼 KLF14 多型性與空腹 insulin 上升、腰臀比、T2D 關聯。
- 機轉:脂肪由 gynoid(女性型)重新分布至內臟、脂肪細胞變大。
- in vitro:透過 preadipocyte 增殖增加但 lipogenesis 受損。
- 因母系印記,效應僅在從母親遺傳給女兒時表現。
GWAS 鑑定的糖尿病基因|Diabetes Genes Identified by Genome-Wide Association Studies
- GWAS 持續找出風險變異並描繪 T2D 遺傳結構。
- 圖 33.3 呈 T2D 與 5 個代謝特徵(BMI、腰圍、腰臀比、空腹 insulin、空腹 glucose)的位點交集,重疊明顯但非完全。
- CREBRF(幾乎只見於薩摩亞人):每 risk allele 增 BMI 1.4 kg/m²,但 T2D 反 odds ratio 0.6。
- 機轉未明,可能脂肪集中於代謝健康的皮下組織而非促炎內臟組織。
- 對 T2D 遺傳的綜合結論:
- 各個基因僅小幅增加風險(OR 1.10–1.45)。
- 多個 at-risk 多型性同存則風險顯著增加 → 衍生多基因 GRS(genetic risk score)。
- 早期 GWAS 偏向胰島素分泌相關基因;近年 SNP 與胰島素敏感性下降相關者增加,部分效應獨立於肥胖。
- 多數風險 SNP 位於非編碼區;部分位於 stretch enhancers(細胞型特異、靠近細胞特化基因)。
- 大量基因關聯但僅解釋 ≤ 10% 遺傳風險;對 missing heritability 探討:
- rare variant hypothesis:常見疾病由多個少見、效應大的變異致病。
- T2D 可分多個表型亞群、各有遺傳特徵,先前混合分析難辨。
- 高家族風險可能來自共享環境、表觀遺傳,或基因結構特徵。
表觀遺傳的糖尿病風險|Epigenetic Risk of Diabetes
- 除基因序列變異外,表觀遺傳標記亦影響基因表現與 T2D 風險。
- 子宮內過度或不足營養可建立有害表觀遺傳標記。
- 荷蘭飢餓冬天(1944–45 年荷蘭,每日 400–800 kcal)後代:較同性別兄弟姊妹(受孕於正常糧食期間)更易肥胖;表觀遺傳標記持續 60 年。
- 全球多次饑荒重現此現象;小鼠模型確認母→子代肥胖風險,並可透過男性後代精子表觀遺傳改變傳至再下一代。
- 妊娠期過度營養同樣造成跨代肥胖與 T2D 風險。
- 父系暴露亦可傳遞。
- 確切機轉與對 T2D 的影響仍研究中。
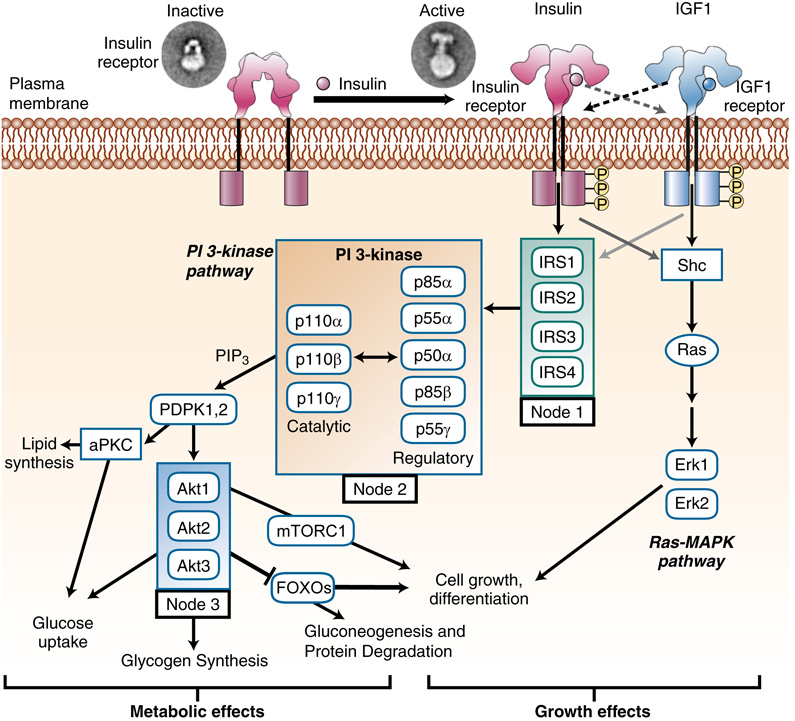
胰島素訊息|Insulin Signaling
- 胰島素訊息透過細胞表面 tyrosine kinase 受體啟動。
- 連鎖磷酸化/去磷酸化、二級訊息、蛋白–蛋白互動 → 全身近乎所有組織的代謝事件。
- Insulin receptor ubiquitously 表達,但作用組織特異(取決於下游目標組成)。
- Insulin receptor 結構:
- 兩個 insulin-binding α-subunit + 兩個催化 β-subunit。
- 由單鏈 prereceptor 切割產生 α₂β₂ 異四聚體。
- Insulin 結合 α-subunit → 啟動 β-subunit 細胞內 tyrosine kinase domain → 磷酸化 intracellular partner 的 tyrosine。
- 冷凍電子顯微鏡:受體外結構由 inverted V → T 形構象變化,使跨膜與細胞內域接近 → trans-phosphorylation 與活化(圖 33.4)。
- 受體磷酸化後,多種 adapter 蛋白(IRSs、Shc、Grb 等)結合並傳訊:代謝調控、有絲分裂、受體內化。
Insulin Receptor 磷酸化後的下游事件|Downstream Events After Insulin Receptor Phosphorylation
- IRS、PI3K、AKT(即 protein kinase B)為 insulin signaling 三個關鍵節點,主導代謝與轉錄效應(圖 33.4)。
- IRS 蛋白
- 既為 substrate 亦為 adapter;多功能域(PH、PTB、SH)使其與 IR 與 IGF1R 的磷酸 tyrosine 互動。
- 多個 tyrosine 位點供下游分子(如 PI3K)對接。
- 四種 isoform(IRS1–IRS4),IRS1 與 IRS2 ubiquitously 表達。
- 功能部分重疊但組織特異:
- 小鼠 IRS1 失活:輕度胰島素阻抗 + 生長遲滯。
- IRS2 失活:β 細胞失能 + 繼發胰島素阻抗。
- IRS 磷酸化後 → 結合並活化 PI3K → 產生 PIP3。
- PIP3 → 活化 PDPK1/2 → AKT isoforms、atypical PKC、wortmannin-sensitive insulin-stimulated serine kinase 等。
- AKT 三個 isoform,需特定 threonine(T308 by PDPK1)、serine(S473 by mTORC2)磷酸化才完全活化。
- AKT 磷酸化下游分子調控脂質合成、肝糖合成、蛋白合成、apoptosis。
- AKT1 失活 → 生長遲滯;AKT2 失活 → 胰島素阻抗 + diabetes(小鼠)。
- 人類胰島素阻抗骨骼肌中 IRS-associated PI3K 與 AKT 活性下降;部分 PI3K 活性下降者,AKT 活化卻正常。
- FOXO transcription factors
- AKT 下游目標。
- AKT 磷酸化 FOXO 三個位點 → nuclear exclusion → 抑制基因轉錄。
- 肝臟:FOXO 透過促 G6Pase 與 PEPCK 轉錄推動糖質新生;亦結合 Sin3a 抑 GSK。
- 肌肉:FOXO 缺失可救回 streptozotocin 糖尿病或 IR/IGF1R 雙剔除所致肌肉萎縮。
- 故代謝組織內 FOXO 缺失可全部或部分救回 T1D 與 T2D 的不利效應。
胰島素的組織特異作用|Tissue-Specific Actions of Insulin
肌肉與脂肪的 insulin-mediated 葡萄糖攝取機轉|Mechanisms of Insulin-Mediated Glucose Uptake in Muscle and Fat
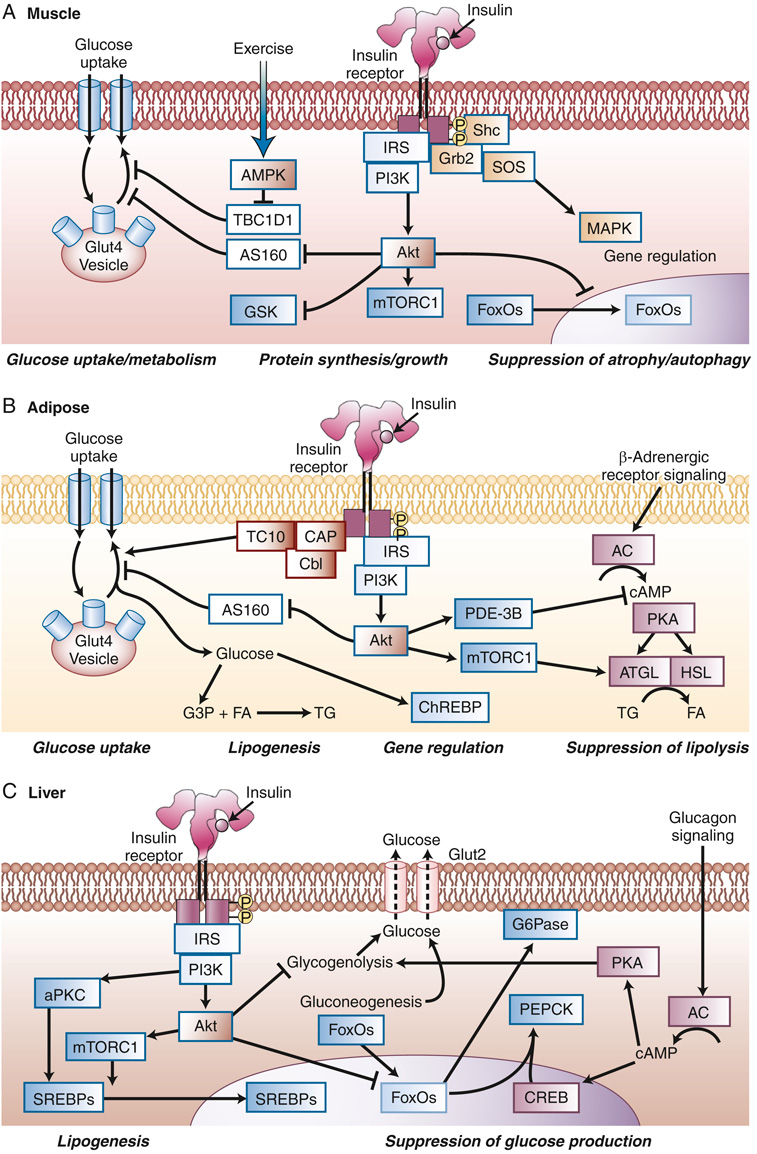
- 胰島素於骨骼肌與脂肪組織主要效應:刺激 GLUT4 由細胞內庫位移位至細胞表面 → 葡萄糖攝取(圖 33.5A)。
- GLUT4 在肌肉或脂肪剔除即造成胰島素阻抗表型。
- AS160(TBC1D4)與 TBC1D1 為 Rab GAP 蛋白,預設抑制 GLUT4 移位;insulin → AKT 磷酸化 AS160,解除抑制,移位 GLUT4 囊泡至膜。
- TBC1D1 亦受 AMPK 磷酸化 → 介導運動誘發肌肉葡萄糖攝取(在 T2D 病人中保留)。
- 胰島素阻抗者骨骼肌 GLUT4 數量未變,但 insulin 啟動移位的能力受損。
- 脂肪組織中還有 PI3K 獨立路徑:IR → CAP–Cbl → TC10(actin remodeling enzyme)→ GLUT4 移位;骨骼肌類似機轉透過 Rac。
Proteostasis
- Insulin 對蛋白恆定(proteostasis)影響深遠。
- 肌肉:促進蛋白合成 + 抑制蛋白降解(proteasome、autophagy)→ 肥大(圖 33.5A)。
- 透過 AKT 活化 mTORC1 → 蛋白合成、細胞生長。
- 同時抑制 FoxO(蛋白降解的關鍵調控者)。
- 小鼠肌肉特異 IR 或 IR + IGF1R 剔除 → 嚴重肌萎縮,FOXO 共剔除可完全救回。
- T1D 動物模型與停用 insulin 8 小時的 T1D 病人皆可見蛋白降解上升。
- 結論:insulin 協調肌肉蛋白週轉,依賴抑制 FOXO 介導的蛋白降解。
脂解與脂肪生成的胰島素調控|Insulin Regulation of Lipolysis and Lipogenesis
- 肝臟為全身 de novo 脂肪生成的主要場所;脂肪組織為三酸甘油酯主要儲存庫。
- 胰島素於脂肪:增加 TG 合成 + 抑制脂解(圖 33.5B)。
- 增加葡萄糖攝取 → glycerol-3-phosphate → 與脂肪酸合成 TG。
- 增加 FA 進入:透過 FATP1、CD36 移位,或增加 LPL 表達/活性。
- 葡萄糖亦活化 ChREBP 上調脂肪細胞糖解與脂肪生成基因。
- 過表達 GLUT4 於脂肪可抗 diet-induced 胰島素阻抗,但若刪除 ChREBP 則此保護失效。
- 抑制脂解(多層次)
- β-adrenergic 訊息 → cAMP → PKA → 活化 ATGL 與 HSL(兩主要脂肪細胞脂解酶)。
- Insulin 活化 phosphodiesterase 3B → 降低 cAMP → 抑制 PKA → 抑制 ATGL/HSL。
- 另透過降低 ATGL mRNA 與增加 FSP27(脂滴穩定蛋白)。
- 臨床意義:未控制 T1D 因脂解未抑 → 脂肪酸氧化 → ketone body 升;新診斷 T1D 開始 insulin 治療第一年常顯著增脂。
- 脂肪分布調控
- GWAS:周邊脂肪減少 + 嚴重胰島素阻抗 + 53 基因 SNP 關聯,含 INSR、IRS1、PIK3R1。
- 與 familial partial lipodystrophy type 1(FPLD1)高度相關。
- 嚙齒類脂肪細胞 IR 剔除 → 脂肪組織快速減少、脂肪重分布至肝、lipodystrophic 糖尿病。
- 肝臟脂肪生成
- SREBP 為脂肪生成基因(FASN、LPL、ACC、SCD)總調節者。
- aPKC λ/ζ → 上調 SREBP1-c 表達。
- SREBP1-c 需蛋白水解切割入核 → mTORC1 介導(位於 IRS-PI3K-AKT 下游);mTORC1 還能穩定 SREBP1 標的 mRNA。
- 小鼠與人 IR 突變 → 肝胰島素訊息減 → 抗肝脂肪變。
- 弔詭:T2D + 代謝症候群者,雖肌肉與脂肪胰島素阻抗,但肝脂肪生成上調 + dyslipidemia;可能來自 pathway-selective postreceptor 阻抗 + altered substrate supply + extrahepatic signaling。
肝糖代謝的胰島素調控|Insulin Regulation of Hepatic Carbohydrate Metabolism
- 肝糖製造由 insulin(抑制)vs glucagon(活化)平衡,insulin 主導(圖 33.5C)。
- 交感神經系統與葡萄糖自我調節亦參與,但較次要。
- 胰島素降低肝糖輸出維持空腹血糖正常與葡萄糖耐受性正常。
- 抑糖原分解(glycogenolysis):insulin 主導 glucagon。
- Glucagon 透過 PKA cascade 增加 cAMP 啟動糖原分解 + 透過 CREB 增加 PEPCK 轉錄促進糖質新生。
- cAMP 觸發機轉複雜:
- CREB 共活化子 TORC2 對 cAMP 反應去磷酸化 → 入核 → 啟動 CREB 依賴的糖質新生酶轉錄。
- CREB 上調 PGC1A → FOXO1 共活化 PEPCK、G6P 等糖質新生基因。
- FOXO 在 IR 剔除小鼠中刪除可使血糖回正常。
- 去乙醯化亦調控糖質新生:
- SIRT1 去乙醯 PGC1A;HDAC + sirtuins 去乙醯 FOXOs → 入核 + 與 HNF4A 互動。
- PGC1A/FOXO1/HNF4A 複合物為強力糖質新生轉錄活化子。
- 胰島素降低內生糖製造的直接與間接機轉:
- 直接:portal insulin 抑糖原分解 + 抑糖質新生(圖 33.5C)。
- 間接:
- 抑制 α 細胞 glucagon 分泌(systemic + paracrine)→ 降低糖原分解與糖質新生啟動。
- 降糖質新生底物供給:抑肌蛋白降解產生的 alanine、抑脂解產生的 glycerol 與 FFA(圖 33.5A,B)。
- 高 insulin clamp 下若以 triglyceride/heparin 維持高 FA,則 insulin 抑肝糖輸出受損。
- glucagon 抑制 + 底物減少 → 加成抑制肝糖產生。
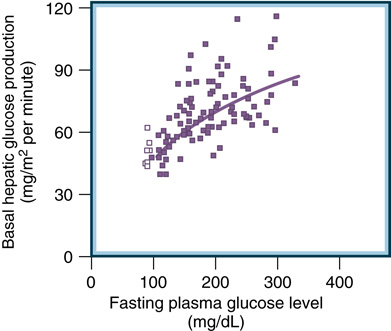
- 臨床:
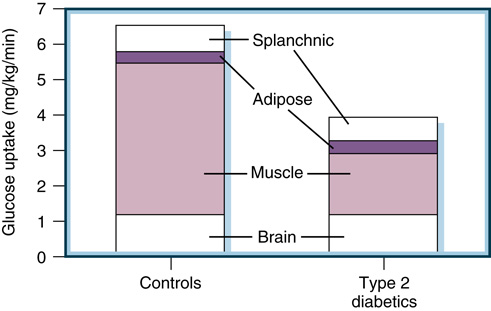
- 肝胰島素阻抗對 T2D 空腹高血糖貢獻很大;其數量級與周邊葡萄糖處置缺陷相當或更大。
- 直接抑制糖原分解的 dose-response 曲線右移。
- 肝糖輸出與空腹高血糖直接關聯(圖 33.6)。
- T2D 中 insulin 抑制肝糖輸出在低與高 insulin 皆受損;早期病程肝糖製造已升高,但較瘦、相對胰島素敏感的 T2D 可能正常。
- Metformin 抑制肝糖製造 → 改善葡萄糖耐受。
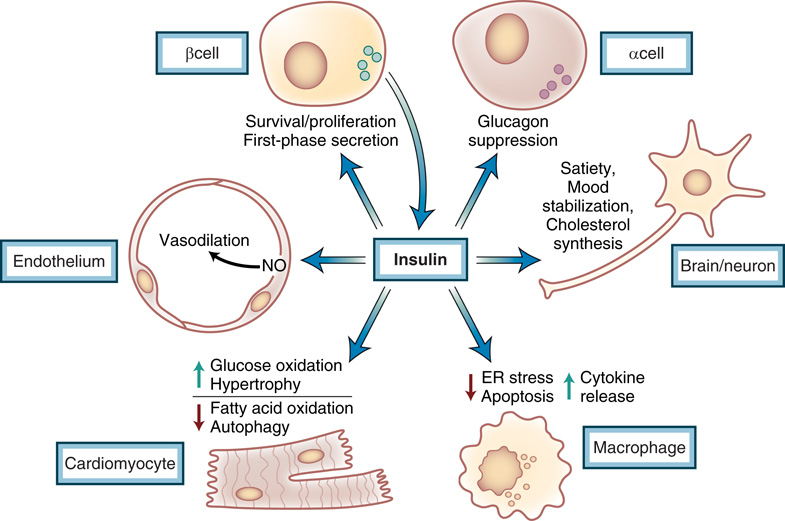
不斷擴展的 insulin-sensitive 組織清單|Expanding Collection of Insulin-Sensitive Tissues
- 雖代謝研究多聚焦於肌肉、肝、脂肪,但 insulin receptor ubiquitous 表達;非經典組織的 insulin 作用對健康亦重要(圖 33.7)。
- β 細胞
- Insulin signaling 對 β 細胞存活與增殖關鍵。
- 慢性輕度高血糖會增加 insulin 分泌與 β 細胞質量,β 細胞質量上調能力影響 T2D 發展。
- β 細胞 IR 剔除 → 失去第一相 insulin 分泌、無法在 diet-induced obesity 與肝胰島素阻抗下擴增 → autocrine loop。
- FOXO 為下游目標,維持 β 細胞功能與身分認同;FOXO 缺失導致 β 細胞去分化。
- 人類胰島素阻抗與 T2D 中 β 細胞去分化漸有證據;對 β 細胞失能貢獻仍研究中。
- α 細胞
- α 細胞 IR 剔除小鼠 → 輕度葡萄糖耐受不良、glucagon 升、漸進式高 insulin 血症(皆代謝症候群與 T2D 特徵)。
- 故 α/β 細胞 insulin signaling 對代謝健康重要。
- 血管內皮
- 過去視為 insulin-insensitive,現知具反應性。
- Insulin → 內皮 NO → vasodilation;胰島素阻抗者此反應受損。
- 內皮 insulin 敏感性對部分周邊組織的 insulin 跨內皮運輸亦重要。
- 不同組織因 IR 密度與毛細血管 fenestration 差異,血管壁通透性影響 insulin 抵達各組織的時程。
- 內皮 IR 剔除小鼠在 atherogenic 模型中 atherosclerosis 病灶增 2–3 倍;過表達 IRS 或抑制 FOXO 改善內皮並預防 atherosclerosis。
- 心肌
- Insulin signaling 控制生長與代謝(生理與病理狀態)。
- 心臟可使用多種底物:禁食偏好 FA。
- Insulin 刺激下 → 葡萄糖氧化增加 + FA 氧化抑制;胰島素阻抗或糖尿病狀態此 metabolic flexibility 喪失。
- Insulin 透過抑 autophagy 促進產後心肌生長。
- 組織交流被破壞 → 代謝症候群 → 心血管風險增加。
- 巨噬細胞
- 巨噬細胞 IR 失能 → 動脈硬化病灶大小略小 + necrotic core 惡化(ER stress 增 apoptosis)。
- 此觀察延伸至肥胖與老化模型 → 巨噬細胞胰島素阻抗在代謝症候群下推進斑塊進展。
中樞神經系統的 Insulin Signaling|Insulin Signaling in the Central Nervous System
- 過去認為 insulin-independent,現知腦為 insulin signaling 重要場所。
- 腦多數區域以 GLUT1、GLUT3 攝糖(insulin 獨立),但 IR 在腦廣泛表達。
- 小鼠腦特異 IR 剔除 → 體重增、低性腺刺激素性低性腺、肝糖製造增、抑鬱表型。
- 臨床:腦胰島素阻抗與內臟脂肪相關;腦阻抗較輕者,生活型態介入反應較佳。
- 細胞型特異效應
- 神經元 IR 為調控肝糖製造的關鍵。
- 星狀細胞 IR 透過 ATP 分泌影響情緒。
- IR 活化下游:膽固醇合成 → 突觸傳遞;MAPK → 增殖;GSK3B → 軸突生長與神經可塑性;FOXO → 基因轉錄、神經元極性;mTORC1 → autophagy、蛋白合成、神經可塑性。
- Insulin 進入腦具區域差異,部分依 receptor-mediated transcytosis;下視丘 tanycytes 的 IR 控制 hypothalamic insulin 攝入與 AgRP 神經元活性、食慾。
- 全身胰島素阻抗 → CSF insulin 較期望低(BBB 運輸下降)。
- 腦特異 insulin + IGF1 訊息阻抗與 Alzheimer disease 關聯;對病程進展的意義仍未定。
- IR 兩個 isoform:IR-A 缺 exon 11,IR-B 含;IR-A 對 IGF2 親和力較高。腦主要表達 IR-A。
- 腦 IR 獨特糖化模式 + 與 IGF1R 形成 heterodimer(偏 IGF1 訊息)。
- 鼻內 insulin 對 Alzheimer disease、抑鬱、肥胖試驗中。
胰島素阻抗與第 2 型糖尿病風險|Insulin Resistance and the Risk of Type 2 Diabetes Mellitus
胰島素阻抗|Insulin Resistance
- 定義:對外源或內源 insulin 的生物反應受損。
- 主要表現:
- 骨骼肌 insulin-stimulated 葡萄糖運輸與代謝降低。
- Insulin 抑制脂肪細胞脂解能力下降。
- Insulin 抑制肝糖輸出能力下降。
- 因 insulin 多效作用,阻抗可導致多器官系統與胺基酸、葡萄糖、脂質代謝異常。
- 測量方法
- 黃金標準:hyperinsulinemic euglycemic clamp。
- 持續輸 insulin 達高 insulin 狀態,同時調整 glucose 輸注維持 euglycemia。
- 穩態下 glucose 輸注速率 = 葡萄糖攝取/處置速率 = insulin 敏感性。
- 加 tracer 可測肝糖製造率(肝 insulin 敏感性)。
- Reaven 的 SSPG(steady-state plasma glucose)法。
- Bergman 的 minimal model(FSIVGTT)。
- Disposition Index:結合 insulin 敏感性與分泌的綜合測量。
- HOMA-IR:用 fasting glucose + insulin 計算,與 clamp 相關性 R_s = 0.88。
- QUICKI:HOMA-IR 倒數對數,方向相反但類似。
- 黃金標準:hyperinsulinemic euglycemic clamp。
- 影響 insulin 敏感性的因子:年齡、體重、族群、體脂(特別是腹腔內肥胖)、運動、飲食、菌群、藥物。
- 胰島素阻抗於 T2D 一致存在,且早於糖尿病多年。
- 前瞻研究:胰島素阻抗早於並預測糖尿病發生。
- 環境因子對遺傳易感性影響強。
- 雖肥胖與胰島素阻抗整體相關,但即使 BMI > 25 或 30 kg/m²,個體間敏感性變異仍大。
- 分子機轉
- Insulin receptor β-subunit 的 serine/threonine 磷酸化降低受體自磷酸化。
- 多種抑制性 Ser/Thr kinase 在動物模型與人胰島素阻抗中升高。
- 介入降低受體 serine 磷酸化 → 增 insulin signaling。
- PTP1B、LAR 等 protein tyrosine phosphatase 增加去磷酸化、削弱訊息;二者於胰島素阻抗者升高。
- PTP1B 剔除小鼠:insulin 敏感性增、抗 diet-induced obesity。
- T2D 病人 iPS 衍生肌肉細胞具多重 insulin signaling 異常 → cell-autonomous + extrinsic 因子並存。
- 罕見型胰島素阻抗:
- IR 突變相關之罕見阻抗(前述)。
- IR autoantibodies(type B insulin resistance + acanthosis nigricans):嚴重阻抗,常需數千單位/日 insulin;可用 combined immunotherapy 治療。
肥胖與第 2 型糖尿病|Obesity and Type 2 Diabetes Mellitus
- 肥胖與 T2D 關聯數十年來已被確立(圖 33.9)。
- 中央型(intra-abdominal、visceral)肥胖比總脂肪更強烈關聯:
- 胰島素阻抗。
- 肝、肌肉的 ectopic fat。
- 升血糖、升 insulin。
- 升總膽固醇與三酸甘油酯。
- 降 HDL-C。
- 部分研究:皮下脂肪可能對胰島素阻抗具保護性。
- 故腹部脂肪與葡萄糖耐受性的關聯獨立於總體脂肪。
- 機轉假說(可能交互作用):
- 腹部脂肪脂解活性高(adrenergic 受體較多)。
- 對 insulin 抗脂解效應較具阻抗(含 LPL 活性改變)→ 脂解增 → FA 入循環(門靜脈承受最大)。
- 皮下脂肪生成更多 adiponectin(有益脂肪激素)。
- 腹部脂肪 11β-HSD1 高 → cortisone → cortisol 局部升 → 改變脂肪細胞脂解與脂肪激素分泌 → 直接調節葡萄糖代謝。
- 脂肪組織分泌外泌體(exosomes),被其他組織攝取調控基因表現,亦促胰島素阻抗。
高胰島素血症與胰島素阻抗|Hyperinsulinemia and Insulin Resistance
- 高 insulin 本身可致胰島素阻抗。
- 升高 insulin → 下調 IR、postreceptor pathway desensitization。
- Del Prato 等:正常人持續 24/72 h 生理性高 insulin → 抑 nonoxidative 葡萄糖處置 + glycogen synthase 啟動受損。
- 抑制肥胖、阻抗者的 insulin 分泌 → 增 insulin 敏感性。
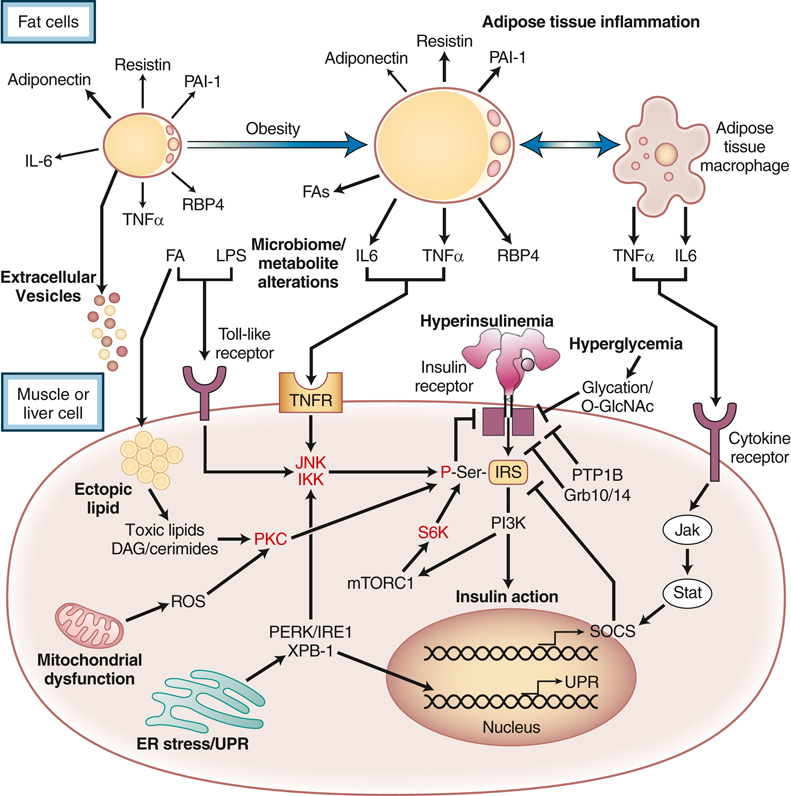
- 細胞層次:
- IRS serine 磷酸化抑制 insulin signaling,由多種 kinase 介導,包含 mTOR/S6K(PI3K-AKT 下游)。
- 適當位點的 IRS serine 磷酸化 → 增 ubiquitination 與降解 → insulin signaling 下降。
- 然 in vivo 較複雜:將 IRS1 S302A 突變的小鼠並未防止 mTOR 活化引起肝胰島素阻抗。
- mTOR 透過磷酸化增加 GRB10 → GRB10 與 GRB14 為 insulin signaling 負調節器(肌肉與脂肪),其多型性與 T2D 相關。
- ERK(insulin → MAPK 下游)反饋抑制 IRS。
- FOXO 轉錄調控 IR 與多種 AKT 抑制因子(TRB3、PP2A)。
- 結論:insulin signaling 自我反饋;高 insulin 慢性活化即透過 feedback 抑制 insulin signaling。
營養過載與胰島素阻抗|Nutrient Overload and Insulin Resistance
- 細胞具多種感測營養機轉:直接與間接活化轉錄因子與 protein kinase。
- 胺基酸 → mTORC1 → 生長。
- 脂肪酸:TLR、PPARα、PPARγ。
- 葡萄糖 → O-GlcNAC 後轉譯修飾 Ser/Thr,與磷酸化類似或對立。
- 多數訊息對 IR 訊息提供反饋抑制(透過 Ser/Thr kinase 如 S6K、JNK、IKK)。
- 慢性過載扭曲正常細胞訊息 → 胰島素阻抗(圖 33.8)。
- 換言之:對肥胖的組織反應原為對營養過剩的正常生理反應,長時間活化卻變成胰島素阻抗、發炎、細胞死亡。
脂肪組織與胰島素阻抗|Adipose Tissue and Insulin Resistance
- 過量營養超出消耗 → 增加細胞質量或儲存。
- 不論碳水、蛋白、脂質,最終以 TG 儲於白色脂肪。
- 脂肪儲存能力超載 → 脂質與其他營養進入非儲存組織 → ectopic lipid 累積(肌、肝、血管、β 細胞)→ 毒性脂質代謝物(DAG、ceramide)→ 活化 PKC isoforms → 胰島素阻抗。
- 脂肪擴張能力不足在過量飲食時亦促胰島素阻抗。
- 脂肪細胞不只是儲存:
- 調控 FA 攝取/釋放、glycerol-FA cycle。
- 釋出 leptin 等能量訊息激素。
- 分泌愈來愈多 cytokine(hormonal、paracrine、autocrine)。
- 肥胖時脂肪細胞表面積增加 → leptin、IL6、IL8、MCP1、G-CSF 增表達 → 招募 M1 促炎巨噬細胞 → 釋出 TNFα 等 → 局部與全身發炎。
- BAT(棕色脂肪)
- 含 multiloculated 脂滴、富粒線體、UCP1 表達 → 解偶聯電子傳遞 → 產熱。
- 受交感神經活化 → FA 動員與氧化。
- 隨年齡與肥胖負相關,可能是阻抗的因或果。
- 50,000+ 人 PET 分析:active BAT 較高者 T2D、dyslipidemia、CVD 較少。
- 長期或反覆冷暴露可增頸與鎖骨上 BAT 質量與活性,改善葡萄糖恆定。
- 米色(beige/brite)脂肪細胞混雜於白脂中,受冷或激素刺激顯現;亦表達 UCP1 但發育起源不同。
- 人類能否擴增 beige、影響代謝度仍未明,但具治療標的潛力。
異位脂肪累積|Ectopic Lipid Accumulation
- 脂肪組織儲存能力超載 → 脂質累積於肌、肝(不適合儲脂的組織)→ 代謝功能異常。
- 肌肉
- Insulin-stimulated 葡萄糖攝取與肌內 TG 量呈反比(biopsy / CT / MRI 可區分肌內 vs 肌外)。
- T2D 一級親屬肌內脂肪增加,與胰島素阻抗相關。
- 機轉:肥胖、胰島素阻抗者肌內 TG 增加可能因 FA 攝取與氧化失衡。
- 靜息餐後 30% FA 氧化、70% 再循環為 TG → 顯示生理儲備超過即時氧化需求。
- FA 攝取、運輸、代謝高度調節;肌肉內失衡 → 肌內 TG 增。
- 肥胖增脂解 → 大量 FA 至肌肉 → DAG 累積 → 活化 novel PKC → 反饋於 insulin signaling。
- 肌內 TG 並非必然與胰島素阻抗綁定:
- 運動訓練增加肌內 TG,但同時增加胰島素敏感性與 FA 氧化能力。
- 機轉:訓練者肌肉中 PLIN2、PLIN3、PLIN5 perilipin 蛋白快速與脂滴關聯 → 改善脂質儲存、隔離脂質中介物 → 防 FA 引起阻抗(亦見於急性運動)。
- 肝
- 脂質累積 → MASLD(先前 NAFLD)。
- 胰島素阻抗下底物供肝量增加為關鍵:
- 脂肪組織脂解 → FA 增加。
- 肌肉胰島素阻抗 → 餐後高血糖 → 肝過量底物 → acetyl-CoA + 毒性脂質中介物。
- Lipodystrophy 人與小鼠模型:無脂肪儲存能力 → 肝代謝失能(MASLD + 嚴重胰島素阻抗)。
- 過量葡萄糖或 fructose 飼小鼠也誘發 MASLD(不只脂質攝取)。
- 結論:多餘巨量營養 + 脂肪儲存力下降 → 肝脂質累積與胰島素阻抗並存。
內質網壓力 / 未摺疊蛋白反應|Endoplasmic Reticulum Stress/Unfolded Protein Response
- ER 負責後轉譯處理(蛋白摺疊、成熟、品管、運輸)。
- 未摺疊蛋白累積 → 啟動 UPR:減慢蛋白合成 + 增加 chaperone 與處理保真度蛋白。
- 三條主要 UPR 路徑:
- PERK → 磷酸化 eIF2α → 抑制多數蛋白合成。
- IRE1 → 切割 XBP1 mRNA → 活化 XBP1 轉錄因子。
- ATF6 + XBP1 → 啟動 chaperone、ER biogenesis、磷脂合成、分泌相關基因。
- 過量飲食與肥胖時,肝、脂肪、β 細胞、肌肉等可活化 UPR:
- 活化 JNK 與 NF-κB / IKK 路徑 → 降 IRS1 活性 + 增促炎介質 → 胰島素阻抗。
- 改變 SREBP1 介導轉錄、降低肝糖質新生。
- 長期活化 → 細胞失能與凋亡。
先天免疫|Innate Immunity
- 先天免疫原認為區別自我 vs 非我,現知為對細胞壓力的整體反應,活化發炎與細胞修復。
- 包含 PRR(pattern recognition receptors):TLR、C-type lectin。
- 表達於巨噬細胞、單核球、樹突細胞、嗜中性球、上皮細胞、適應性免疫細胞,多浸潤肥胖者脂肪。
- 偵測微生物相關脂質、核酸 → NF-κB + AP1 轉錄 → 升 cytokines/chemokines。
- 啟動 inflammasome(多單元複合體)→ 控 caspase 1 介導 IL1β 後轉譯成熟與分泌 → 強烈促炎 → T2D 風險(透過 insulin 阻抗、β 細胞失能)。
- 急性感染與肥胖中先天免疫活化伴顯著胰島素阻抗。
- 部分由升高的 FA 介導:TLR2/4(原應對細菌細胞壁脂質)也被飽和 FA 活化。
- 多元不飽和 FA 抑 TLR signaling。
- 肝細胞、肌肉、脂肪細胞 TLR 活化 → 胰島素阻抗;TLR4 剔除小鼠對 FA 引起阻抗具保護。
粒線體異常|Mitochondrial Abnormalities
- 胰島素阻抗、肥胖、T2D 中皆可見氧化能力下降(人與動物)。
- 肌內脂肪增加可能因粒線體質量變化。
- 一研究:T2D 父母的胰島素阻抗年輕後代 → insulin-stimulated 肌肉葡萄糖攝取下降 60%、肌內脂質增 80%、粒線體氧化能力降 30%;伴隨 type I(氧化)肌纖維比例下降。
- PGC1A、PGC1B 對粒線體生合成與發育中肌纖維型選擇重要:
- 肥胖伴 IGT 與 T2D 者表達下降。
- 電子傳遞鏈活性減、肌纖維內粒線體較小,與阻抗嚴重度相關。
- 育種低有氧能力鼠:肥胖、阻抗、高血壓、dyslipidemia、PGC1A 下降,類似人。
- 但小鼠肌肉過表達 PGC1A 並未改善高脂飲食代謝 → 增粒線體量未必有益。
- 部分研究質疑粒線體異常為阻抗的「因」:
- Insulin 自身可上調粒線體生合成;T1D 撤 insulin → 粒線體基因轉錄與 ATP 合成下降。
- IR 或 IR/IGF1R 雙剔除小鼠肌肉:粒線體呼吸、ATP 製造、TCA / OXPHOS 基因表現均降,FOXO1/3/4 共剔除可救回。
- ATP 周轉減少(久坐)→ 降 ADP/ATP → 抑 ETC → NADH 升 → 抑 TCA → 增 ROS。
- 高脂飲食小鼠:TCA 中介物減 + β 氧化不足 → 偶數鏈 acyl-carnitine 升;肥胖、阻抗者血漿 acyl-carnitine 升為阻抗 marker。
- 粒線體蛋白後轉譯修飾(lysine 乙醯化、succinylation、malonylation)影響活性:
- 一般降酶活性,無酶催化的乙醯化(pH 依賴非酶反應,acetyl-CoA / acyl-CoA 為供體)。
- 粒線體營養通量增 → 乙醯化增 → 抑進一步利用。
- SIRT3 為粒線體主要去乙醯化酶,由 NAD⁺ 活化,去乙醯肝 LCAD 與肌肉 PDH。
- SIRT5 為 NAD⁺ 依賴,去 succinylation/malonylation。
- 運動 → ATP 用量增 → NADH→NAD⁺ 增 → 提升 SIRT3/5 活性 → 增粒線體底物利用能力。
細胞外囊泡與器官間溝通|Extracellular Vesicles in Interorgan Crosstalk
- 細胞釋出多種大小、多種貨物的囊泡 → 進入細胞外或循環 → 被靶組織攝取。
- sEV / exosomes 在肥胖代謝併發症中受最多關注(白脂、棕脂、其他組織皆可釋)。
- 攜帶多種貨物,含大量循環 microRNA(miRNA)。
- miRNA:21–25 nt 非編碼 RNA,結合 mRNA 阻翻譯或促降解。
- 減重後循環 miRNA 模式變化是許多有益效應的關聯。
- sEV 還能含粒線體(脂肪→心交流)、FA 代謝成分(影響 melanoma 病理)。
- 人類:肥胖 + MASLD 病人脂肪組織 sEV 使肌與肝細胞發生胰島素阻抗(AKT 磷酸化下降),未 MASLD 之肥胖或瘦者則無。
骨骼肌胰島素阻抗|Skeletal Muscle Insulin Resistance
- 餐後葡萄糖主要去處為骨骼肌;主要儲存方式為轉換為 glycogen。
- 肥胖中肌肉胰島素阻抗早於脂肪與肝阻抗 → 反映肌肉儲存能力有限。
- Hyperinsulinemic clamp 顯示:阻抗者(含 T2D)非氧化葡萄糖處置缺陷 → 主要因 glycogen 合成缺陷,繼發於 insulin-stimulated 葡萄糖攝取下降(圖 33.10)。
- 升高 FA 預測 IGT → DM 進展;周邊 FA 因肝肌高效萃取可能不顯著升。
- 內臟脂解增加 → FA 至肌 → 抑肌葡萄糖攝取。
- Randle 葡萄糖–脂肪酸週期假說:
- FA 增 → mitochondrial acetyl-CoA/CoA、NADH/NAD⁺ 比上升 → 抑 PDH。
- 細胞內 citrate 上升 → allosteric 抑 PFK → glucose-6-phosphate 累積 → 抑 hexokinase II → 細胞內葡萄糖升、攝取下降。
- 較新人類研究(¹³C / ³¹P NMR):
- 高 insulin 下 FA 主要效應為降葡萄糖運輸,非 Randle 預測。
- 三酸甘油酯 + heparin 輸 → 細胞內葡萄糖與 G6P 下降早於 glycogen 累積下降。
- 反 Randle 預測(其預測 G6P 升)。
- T2D 與其瘦、葡萄糖正常的胰島素阻抗後代亦見類似葡萄糖運輸下降。
- PI3K 活性降低、novel PKC(PKCθ、PKCδ 等)活性升 → 部分介導 FA 引起阻抗。
- PKC 磷酸化 IKKβ → 降解 → NF-κB 入核 → FA 引起阻抗。
- 高劑量 salicylate 干擾 IKKβ → 改善 T2D 的胰島素敏感性與葡萄糖代謝(小型試驗)。
骨骼肌脂肪酸代謝|Fatty Acid Metabolism in Skeletal Muscle
- CPT1 在粒線體葡萄糖–脂肪酸代謝平衡的核心作用受多年關注;malonyl-CoA 抑制 CPT1(肝臟)阻 mitochondrial FA 攝取。
- 肌肉
- 高葡萄糖、高 insulin → 增 TCA flux → citrate efflux 至胞質 → 透過 ATP citrate lyase 轉 acetyl-CoA → ACC(acetyl coenzyme A carboxylase)的底物。
- Citrate allosteric 活化 ACC,催化 acetyl-CoA → malonyl-CoA。
- 人體:高速 insulin + glucose 輸 → 肌 malonyl-CoA 升、全身(推測肌肉)FA 氧化降。
- 但肌肉 CPT1 主要 isoform(97%)對 malonyl-CoA 抑制敏感性低 100 倍 → 提示 CPT1 自身量更重要。
- 嚙齒類肌肉過表達 CPT1 → FA 氧化增、肌內 TG 降。
- CPT1 mRNA 由 PPARA 活化子、脂飼、運動調控;與肥胖呈反比。
- 不論透過 malonyl-CoA 抑制或 CPT1 表達下降,肌肉 mitochondrial FA 氧化受抑 → 毒性脂質中介物(如 DAG)累積 → 活化 novel PKC → 阻抗(圖 33.8)。
- 支持證據:肥胖與 T2D 病人 malonyl-CoA 升、ACC 磷酸化降、FA 氧化下降、肌內 acyl-CoA 與 TG 累積;3 個月 rosiglitazone 治療可逆轉。
- UCP(uncoupling protein)
- UCP1:proton translocator,使呼吸鏈質子梯度散逸 → 解偶聯氧化磷酸化 → 產熱(BAT)。
- 高 active BAT 量者白脂較少、代謝較佳。
- UCP2、UCP3 結構類似 UCP1,但生理條件下是否解偶聯仍未定。
- UCP3 mRNA:主要骨骼肌與 BAT。
- UCP2:ubiquitous,可能與 ROS 解毒、CNS 代謝調控有關。
- UCP2/3 mRNA 表現受甲狀腺素與高 FA 上調。
- 人體:高脂飲食上調 UCP2/3 mRNA,type IIA 肌纖維比例高者尤甚。
- 運動訓練增粒線體氧化能力但不變 UCP2/3 量(小研究)。
- 肥胖與 UCP3 splice isoform 正相關。
- UCP3 啟動子區域多型性與肌肉 UCP3 表達相關。
晝夜節律、肥胖與胰島素阻抗|Circadian Rhythms, Obesity, and Insulin Resistance
- 哺乳類 24 小時晝夜週期由整合的轉錄–翻譯反饋網路控制。
- 核心時鐘基因不僅在下視丘 SCN,也存於幾乎每個細胞。
- 主要轉錄因子:
- CLOCK + BMAL1 形成異二聚體,結合目標基因(PER、CRY)啟動子。
- PER、CRY 累積 → 異二聚體入核 → 抑 CLOCK-BMAL1 轉錄活性(負反饋)。
- 第二負反饋:CLOCK-BMAL1 誘 REV-ERBα(NR1D1)→ 抑 BMAL1 轉錄。
- 後轉譯磷酸化、ubiquitination 進一步調節。
- 流行病學:人類睡眠減少 → 肥胖、T2D 等代謝異常增加(圖 33.11)。
- 實驗性睡眠中斷 → 胰島素作用受損、leptin/ghrelin 改變 → 食慾增、發炎 cytokine 升、心血管風險改變。
- 餵食模式偏離晝夜節律 → 營養出現與代謝酶失同步 → 脂質至易感組織 → lipotoxicity + leptin 降 → 食慾增。
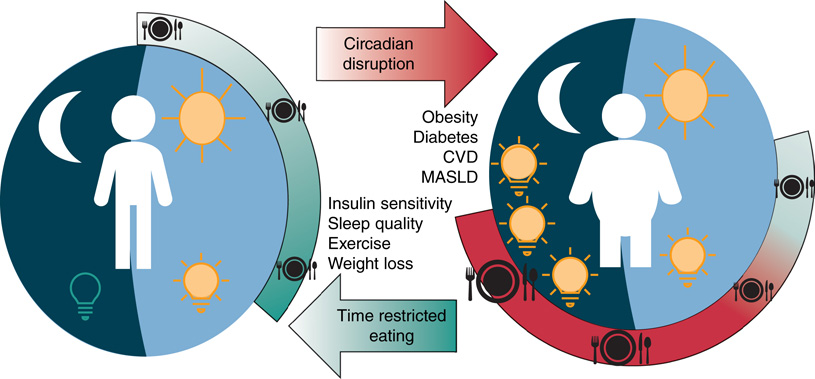
- 阻塞型睡眠呼吸中止(OSA):睡眠破碎 + 缺氧 → 阻抗與糖尿病風險。
- 治療 OSA 可改善 T2D 血糖控制,但治療順從度為主要障礙。
- 限時飲食(TRF / TRE,每 24 h 限 8–10 h 進食)對代謝健康顯著改善(見圖 33.11)。
- 高脂自由餵養小鼠:肥胖 + 阻抗;同熱量限 8 h 進食則無 → 防肥胖、高 insulin、肝脂變、發炎標記。
- 人類:5 週 6 h vs 12 h 等熱量進食 → 改善 prediabetic 男性的胰島素敏感性與血壓,無顯著體重變化。
- 智慧型手機追蹤的 10 h TRE 改善體組成、體重、血糖控制。
- TRE 益處部分需伴隨體重下降;額外加在熱量限制上未必再加分。
腸道菌群與代謝物在糖尿病與胰島素阻抗中的角色|Role of the Gut Microbiome and Metabolome in Diabetes and Insulin Resistance
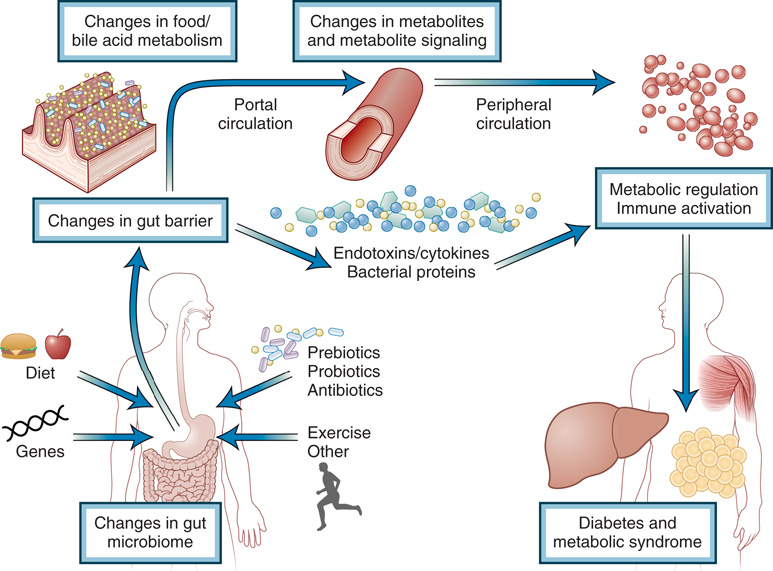
- 飲食與環境因子作用於糖尿病的重要中介為腸道菌群。
- 哺乳類菌群於出生時形成、嬰兒期重塑、成人後相對穩定,但仍可受飲食、抗生素、疾病改變。
- 多為共生(無害)或互利(有益):營養與外源物代謝、維持腸黏膜屏障、防病原、免疫調節。
- 過去十年顯示:菌群可促肥胖、糖尿病、代謝症候群、胰島素阻抗(嚙齒類與人);亦中介遺傳、飲食、減重手術效應。
- 小鼠:早期低劑量抗生素擾亂正常菌群發育 → 易肥胖與葡萄糖耐受不良。
- 機轉(圖 33.12):
- 屏障功能改變、不可消化成分分解、膽酸與其他物質修飾、腸道發育、免疫系統教育。
- 釋出細菌蛋白、內毒素、cytokine 入血。
- 改變數百種代謝物:膽酸、短鏈脂肪酸、胺基酸等。
- 共同 → 組織特異代謝失調 + 免疫活化 → 胰島素阻抗 + 糖尿病進展。
- 與胰島素阻抗相關代謝物(鼠與人):
- SCFA(acetate、propionate、butyrate):腸與代謝健康關鍵。
- 2-aminoadipate、α-hydroxybutyrate、N-acetylglycine 等:T2D 生物標記候選。
- 治療方向:prebiotics、probiotics、糞菌移植,但需更多研究確證。
引發胰島素阻抗的特殊狀況|Special Conditions That Induce Insulin Resistance
妊娠糖尿病|Gestational Diabetes
- 分類:第一孕期診斷 → 視為原存 T2D;第二、三孕期診斷 → 妊娠糖尿病。
- 妊娠糖尿病雖多於分娩後緩解,仍為未來 T2D 的顯著風險因子。
- 妊娠中胰島素阻抗為正常生理:
- 隨孕期進展阻抗加重 → 母體 insulin 分泌增最高至 250% 代償。
- 代償不足 → 妊娠糖尿病。
- 孕前肥胖為主要風險因子。
- 妊娠胰島素阻抗的驅動者非單純體重:分娩後數日血糖即回正常 → 提示胎盤衍生因子主導阻抗與 β 細胞擴增。
- 確切因子難定(人鼠 β 細胞生理差異 + 介入研究困難)。
- 候選:placenta-derived growth hormone(部分貢獻)、placental lactogen、placenta-derived 促炎 exosomes、placenta-derived miRNA。
藥物與壓力誘發的胰島素阻抗|Drug- and Stress-Induced Insulin Resistance
- 多種疾病與藥物可誘發胰島素阻抗或合併分泌減少 → 葡萄糖耐受不良(Box 33.2)。
Box 33.2 與胰島素阻抗相關的藥物與壓力源
藥物:
- Glucocorticoids
- HIV 治療藥物(皆有不同程度代謝異常)
- Calcineurin inhibitors
- mTOR inhibitors
- Phosphoinositide 3-kinase inhibitors
- Statins
- Antipsychotics
壓力源:
- 妊娠
- Glucotoxicity
- 手術
- 發炎(肥胖或感染)
- 過量飲食
糖皮質固醇引起的胰島素阻抗|Glucocorticoid-Induced Insulin Resistance
- Cushing syndrome 與外源 GC 治療皆顯著誘胰島素阻抗。
-
80% 非糖尿病類風濕關節炎病人接受 prednisone > 30 mg/day → HbA₁c 上升。
- 機轉多重:
- 快速效應:肝糖製造增加(透過糖質新生)+ 急性肌肉胰島素阻抗(部分因 IRS1 下調)。
- 組織特異:24 小時 hydrocortisone 反增脂肪 insulin 敏感性,但全身 insulin 敏感性下降。
- 組織交流重要:脂肪組織特異 GR 剔除小鼠對長期 dexamethasone 仍保有部分肌肉 insulin 敏感性。
- 長期 GC:
- 脂肪由周邊重分布至中央。
- 脂解增 → TG 與 FA 升。
- Insulin 分泌降。
- 肌肉蛋白降解增 → 肌萎縮 + 皮膚整合性改變。
- 透過 GC 介導的轉錄修飾 + 抑制 insulin / 生長因子訊息綜合作用。
移植後糖尿病|Posttransplant Diabetes Mellitus
- 器官移植後常見 IGT 或 PTD(與免疫抑制劑相關)。
- 多數病人移植後立刻短暫高血糖(手術壓力 + 大劑量類固醇);故 PTD 診斷應於出院、免疫抑制劑穩定後。
- 共識建議:移植後第一年勿用 HbA₁c 篩 PTD(紅血球週轉變化大易漏診)。
- 用藥組合依器官別,但常含長期 glucocorticoids、mTOR inhibitors(sirolimus、everolimus)、calcineurin inhibitors(tacrolimus、cyclosporine)。
- 機轉:
- GC 與 mTOR inhibitor 主要透過誘胰島素阻抗。
- mTOR inhibitor 直接抑 IR 下游 mTOR complex(圖 33.8);單用未必致 DM,但有風險因子(特別合併 CNI)即可誘發。
- Calcineurin inhibitor 主要降低 insulin 分泌:透過降 insulin 合成與 glucokinase 活性;亦有部分增加阻抗(資料分歧,可能僅在風險者)。
發炎與發炎細胞激素|Inflammation and Inflammatory Cytokines
- 肥胖伴隨脂肪組織發炎、巨噬細胞浸潤、cytokine 表達增加。
- 機轉:
- 人與動物模型:IR 數量與激酶活性下降、IRS1/PI3K/AKT 活化受損。
- 部分由脂肪細胞與浸潤巨噬細胞產生的 TNFα 等驅動。
- TNFα → 大鼠 IR 激酶活性下降;3T3-L1 脂肪細胞中亦然 → 可能透過 JNK/IKK 升 + IR/IRS1 serine 磷酸化升。
- TNFα 基因剔除高脂飼小鼠 + neutralizing antisera / soluble TNF receptor 處理大鼠 → IR 激酶活性升 → insulin 敏感性升。
- 但人類降 TNFα 介入幾無改善肥胖或 T2D 的胰島素阻抗。
HIV 感染|Human Immunodeficiency Virus Infection
- HIV 抗病毒治療使其慢性化,但藥物伴顯著代謝風險。
-
3,800 人 cohort:HIV 病人 T2D 盛行率近兩倍;多由 protease inhibitor 致。
- HIV 病人 AMI 風險升 75%(亦與 PI 用藥相關)。
- 其他心血管風險因子:男性、AIDS 診斷、抗病毒反應、CD4 T cell 增加。
- HIV 抗病毒治療下可見 lipoatrophy 與 lipohypertrophy。
- Lipoatrophy 主要見於老的 thymidine 類似 NRTI。
- Lipohypertrophy 機轉不明。
- 多藥組合與頻繁更動使單藥貢獻難分辨。
- 機轉(Koethe et al. 詳述):
- PI 部分可在體內外抑制葡萄糖運輸(可能透過 GLUT4 互動)→ 特異抑 insulin-responsive 組織葡萄糖攝取。
- HIV lipodystrophy 病人脂肪組織粒線體數與氧化功能下降(HAART)。
- PI 直接影響脂肪細胞分化。
- 治療下脂肪 Dicer(miRNA 處理酶)下調 → 循環 exosomal miRNA 降 → 可能促胰島素阻抗。
- 較新 integrase strand transfer inhibitor 增重,可能透過下視丘增食慾。
- HIV-related lipodystrophy 治療仍不理想;新 PI 可能風險較低。
- 切換離開 thymidine 類似 → 部分改善 lipoatrophy。
- TZD 治療:insulin 敏感性改善但脂肪分布改善有限。
- 心血管風險高 → 必治高血脂;標準心血管風險預測低估 HIV 病人風險。
- Tesamorelin(GHRH analog)核准用於 HIV lipodystrophy 異常脂質:適度重分布內臟脂 + 改善脂質譜。
葡萄糖毒性與葡萄糖胺|Glucotoxicity, Glucosamine
- 高血糖本身可致胰島素阻抗 + β 細胞功能下降。
- Pima 印第安人(全球 T2D 發生率最高之一):空腹血糖與 insulin 敏感性強相關;缺陷主要在骨骼肌;其特殊易感性原因仍未明。
- 葡萄糖代謝物的毒性:
- Glucose 進細胞 → glucose-6-phosphate → 多種命運。
- Hexosamine pathway 為糖解相對少(< 3% 總葡萄糖使用)的支線。
- Hexosamine(如 glucosamine)孵脂肪 → 脂肪細胞胰島素阻抗;於骨骼肌亦如是。
- 大鼠輸 glucosamine → 骨骼肌阻抗劑量依賴性增加。
- 過表達速限酶 GFAT 於肌肉的轉殖鼠 → 嚴重肌肉胰島素阻抗。
- 機轉未明,但 glucosamine 過量會破壞 insulin 介導的 GLUT4 移位。
- Hexosamine pathway 被假設為葡萄糖感測器,使細胞感測並適應現有葡萄糖濃度。
術後高血糖|Postoperative Hyperglycemia
- 手術為顯著壓力(手術傷口、麻醉、疼痛)→ 釋出 cortisol、epinephrine、norepinephrine 等壓力激素。
- 心臟繞道後常用 pressors → 嚴重高血糖。
- 非糖尿病病人術後高血糖預測冠繞與一般手術死亡率。
- 積極治療術後高血糖(糖尿病與非糖尿病皆設目標 140–180 mg/dL)→ 降傷口感染與死亡率。
- Epinephrine 為關鍵介導者(內生或輸入):
- 降 insulin 分泌 + 增肌肉與肝胰島素阻抗(肝糖製造抑制失效)。
- 急症情境下用 epinephrine 輸注 → insulin 輸注速率常需調整。
Statins 與抗精神病藥|Statins and Antipsychotic Medications
- 上述疾病可深刻影響胰島素阻抗與血糖;許多藥物效應較小但於人群層級具意義。
- Statins
- 個別病人血糖變化不顯,但大型試驗 → 服 statin 病人 T2D 風險增加。
- 抗精神病藥
- 多數致增重,部分可至完整代謝症候群 + 糖尿病。
- olanzapine 與 clozapine 風險最高。
- 機轉未明;兒童尤其易增重;終身治療造成長期管理挑戰。
- 鑑於胰島素阻抗與 T2D 多因素本質與其與肥胖的緊密聯繫,其他藥物在合適遺傳背景與環境下也可能增加 T2D 風險。
—— 本章結束 ——
TEACHING SLIDES
Williams Endocrinology 15e Ch33|第 2 型糖尿病的病理生理
01 全球流行病學
- T2D 佔糖尿病 90–95%
- 2021 IDF 估全球 5.37 億,2045 推估 7.38 億
- 美國成人糖尿病 11.3%、糖尿病前期 38%
- 兒童青少年 T2D 16 年相對增加 95%
02 亞裔特殊性
- 較低 BMI 即可發 T2D(內臟脂肪比例高)
- ADA 建議亞裔 BMI ≥ 23 kg/m² 啟動篩檢
- 西化飲食 → 移民與本土皆 T2D 增加
03 四大病理生理核心
- 周邊組織胰島素阻抗(肌、脂、肝)
- 葡萄糖刺激下 insulin 分泌缺陷
- 肌肉葡萄糖攝取下降 → 餐後高血糖
- 肝糖製造增加 → 空腹高血糖
04 額外特徵
- Hyperglucagonemia
- Incretin 異常
- 脂肪細胞脂解加速
- 腎小管葡萄糖再吸收增加
- CNS 代謝調控異常
05 阻抗 vs 分泌缺陷時序
- 胰島素阻抗早於 T2D ≥ 20 年
- β 細胞缺陷於糖尿病前期已可偵測
- 兩者於臨床發病時並存
- 青春期、妊娠、久坐、增重加重阻抗
06 單基因型 IR 突變
- Type A:阻抗 + acanthosis nigricans + hyperandrogenism
- Donohue:嚴重子宮內生長遲滯,2 年內死亡
- Rabson-Mendenhall:身材矮、腹膨、牙甲異常
- 可能需 > 10,000 U/day insulin
07 脂質代謝障礙性糖尿病
- Generalized vs partial lipodystrophy
- LMNA、PPARG、AGPAT2、BSCL2 等多基因
- Generalized:嚴重脂肪肝、leptin 極低、leptin replacement 有效
- Partial:常被忽略,多 leptin replacement 無效
08 多基因型 T2D 遺傳學
-
700 個關聯基因;總體解釋遺傳風險 ≤ 10%
- TCF7L2 為常見變異中 OR 最大者
- KCNJ11/ABCC8 新生兒 DM 可改用 sulfonylurea
- PPARG Pro12Ala 降風險;C-allele 增
09 表觀遺傳跨代效應
- 荷蘭飢餓冬天後代肥胖風險升
- 標記持續 60 年
- 母→子→孫代可遞
- 父系暴露亦可傳
10 胰島素訊息三節點
- 圖:Figure 33.4
- IRS(4 個 isoform)
- PI3K(p85/p55 + p110)
- AKT(3 個 isoform)
- FOXO 為下游主要轉錄調節
11 組織特異作用:肌肉
- GLUT4 移位(AS160 解抑)
- 蛋白合成 mTORC1 增加
- 抑制 FOXO → 抑制蛋白降解
- 運動透過 AMPK→TBC1D1 在 T2D 中保留
12 組織特異作用:脂肪
- 葡萄糖攝取增加
- TG 合成(FATP1、CD36、LPL)
- 抑脂解(PDE3B → 抑 PKA → 抑 ATGL/HSL)
- ChREBP 連結葡萄糖與脂肪生成
13 組織特異作用:肝
- 抑糖原分解 + 抑糖質新生
- aPKC λ/ζ → SREBP1c → 脂肪生成
- 直接:portal insulin
- 間接:抑 glucagon、降 gluconeogenic 底物
14 肝胰島素阻抗
- 圖:Figure 33.6
- 對 T2D 空腹高血糖貢獻量級≥周邊葡萄糖處置缺陷
- 抑糖原分解 dose-response 右移
- Metformin 抑肝糖製造 → 改善 GTT
15 非經典 insulin-sensitive 組織
- β 細胞:autocrine loop、FOXO 維持身分
- α 細胞:抑 glucagon
- 內皮:NO、跨內皮 insulin 運輸
- 心肌:metabolic flexibility
- 巨噬細胞:atherosclerosis 進展
- CNS:肝糖製造、情緒、Alzheimer 關聯
16 胰島素阻抗測量
- Hyperinsulinemic euglycemic clamp(黃金標準)
- HOMA-IR:fasting glu × fasting ins
- QUICKI:HOMA-IR 倒數對數
- Disposition Index:敏感性 + 分泌
- SSPG(Reaven)、Bergman 最小模型
17 高 insulin 自身致阻抗
- 下調 IR + postreceptor desensitization
- mTOR/S6K 反饋抑 IRS
- GRB10/14 負調節
- ERK 反饋
- FOXO 控 TRB3、PP2A
18 中央肥胖優於總脂肪
- 圖:Figure 33.9
- 內臟 > 皮下:與阻抗、ectopic fat 強關聯
- 腹部脂肪脂解高、抗 insulin 抑脂解
- 11β-HSD1 增 → 局部 cortisol 升
- 皮下脂肪可能保護(adiponectin)
19 Ectopic Lipid 累積
- 肌:肌內 TG 與阻抗反比;DAG → novel PKC
- 肝:MASLD(前 NAFLD),底物供肝增加
- Lipodystrophy:無儲存力 → MASLD + 嚴重阻抗
- 過量 glucose / fructose 亦致 MASLD
20 運動與肌內 TG 弔詭
- 訓練者肌內 TG 增加但敏感性改善
- PLIN2/3/5 perilipin 包覆隔離脂滴
- 急性運動亦防 FA 引起阻抗
21 ER stress / UPR
- PERK → 抑蛋白合成
- IRE1 → XBP1 切割
- ATF6 + XBP1 → chaperone、ER 生合成
- 過量飲食活化 UPR → JNK/IKK → IRS1 抑
22 先天免疫與阻抗
- TLR2/4 被飽和 FA 活化
- 多元不飽和 FA 抑 TLR
- Inflammasome → IL1β → β 細胞失能
- TLR4 剔除小鼠抗 FA 引起阻抗
23 粒線體缺陷
- T2D 後代肌肉氧化能力降 30%
- PGC1A、PGC1B 表現下降
- 但因果方向仍辯論
- SIRT3 / SIRT5 去乙醯化關鍵
- Acyl-carnitine 為阻抗 marker
24 細胞外囊泡與 miRNA
- 脂肪 sEV 攜 miRNA 入循環
- 影響肝、肌 insulin signaling
- 肥胖+MASLD 病人 sEV 致他組織阻抗
- 減重改變 miRNA profile
25 骨骼肌阻抗
- 圖:Figure 33.10
- 餐後葡萄糖主要去處 = 肌肉
- 阻抗:glycogen 合成 + 葡萄糖攝取下降
- ¹³C / ³¹P NMR:葡萄糖運輸下降早於 glycogen 累積
- 反 Randle 預測
26 CPT1 與肌肉 FA 代謝
- ACC → malonyl-CoA → 抑 CPT1(肝)
- 肌肉 CPT1 對 malonyl-CoA 敏感低 100×
- CPT1 量本身重要
- DAG → novel PKC → 阻抗
- Rosiglitazone 3 個月可逆轉
27 晝夜節律破壞
- 圖:Figure 33.11
- CLOCK + BMAL1 為主轉錄因子
- PER、CRY 形負反饋
- 睡眠不足、輪班、OSA 為 T2D 風險
- TRE(6–10 h 進食窗)改善代謝
28 腸道菌群與代謝物
- 圖:Figure 33.12
- 屏障、膽酸、SCFA、內毒素血症
- SCFA(acetate、propionate、butyrate)關鍵
- 2-aminoadipate、α-hydroxybutyrate 為 T2D marker
- 治療:prebiotics、probiotics、糞菌移植
29 妊娠糖尿病
- 妊娠胰島素阻抗為正常生理(insulin 分泌增 250%)
- 胎盤 GH、placental lactogen、exosomes、miRNA
- 第一孕期診斷視為原存 T2D
- 未來 T2D 顯著風險因子
30 GC 引起阻抗
-
80% RA 病人用 prednisone > 30 mg/d → A1c 升
- 肝糖製造增 + 急性肌阻抗(IRS1 下調)
- 脂肪:24 h 暫增敏感性
- 長期:脂肪重分布、肌萎縮
31 移植後糖尿病
- 第一年勿用 A1c 篩
- GC + mTOR inhibitor → 阻抗
- Calcineurin inhibitor → 主要降 insulin 分泌
- mTOR inhibitor 單用低風險,與 CNI 合用易致 DM
32 HIV 抗病毒治療
- T2D 盛行率近兩倍
- AMI 風險升 75%(PI 相關)
- Lipoatrophy(thymidine NRTI)vs lipohypertrophy
- Tesamorelin 適度改善脂質譜
33 葡萄糖毒性與術後高血糖
- Hexosamine pathway 過載 → 阻抗
- Pima 印第安人易感
- Epinephrine 為術後高血糖關鍵
- 目標血糖 140–180 mg/dL → 降感染與死亡率
34 Statins 與抗精神病藥
- Statins 增 T2D 風險(個別變化小)
- Olanzapine、clozapine 增重最甚
- 兒童尤易增重
- 多藥組合下風險疊加